We are a computational chemistry research group based at the University of Southampton working under the supervision of Prof. Jonathan Essex.
Our research revolves around the application of the theoretical techniques of statistical thermodynamics and quantum mechanics to the study of organic and biomolecular systems.
Our aim is to rationalise and intepret experimentally observed behaviour at the molecular level, and suggest further lines of experimental inquiry. This work is of direct relevance to rational drug-design and we collaborate extensively with the pharamceutical industry.
Some recent publications and software releases from the group. A full list of publications and software packages are also available.
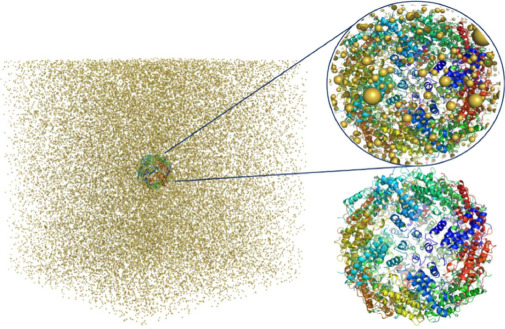
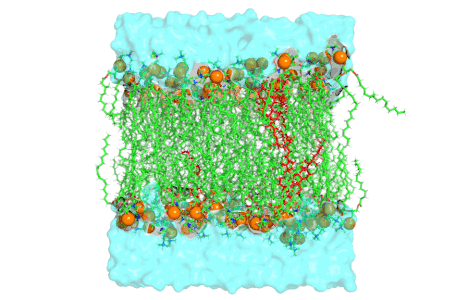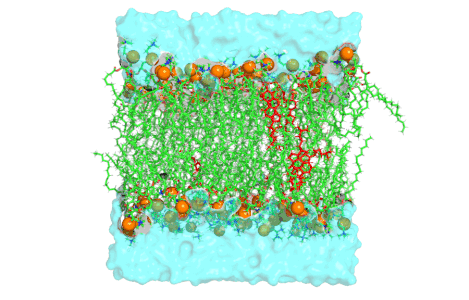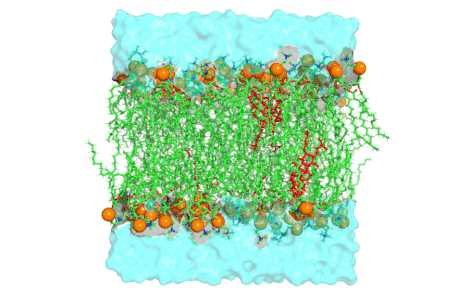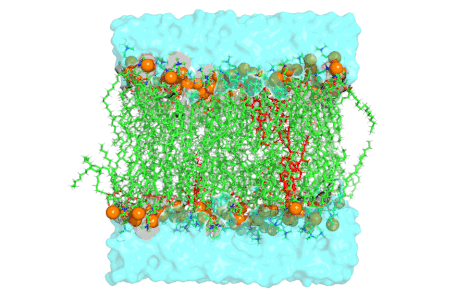
Simulations of cryo-electron microscopy (cryo-EM) images of biological samples can be used to produce test datasets to support the development of instrumentation, methods, and software, as well as to assess data acquisition and analysis strategies. To be useful, these simulations need to be based on physically realistic models which include large volumes of amorphous ice. The gold standard model for EM image simulation is a physical atom-based ice model produced using molecular dynamics simulations. Although practical for small sample volumes; for simulation of cryo-EM data from large sample volumes, this can be too computationally expensive. We have evaluated a Gaussian Random Field (GRF) ice model which is shown to be more computationally efficient for large sample volumes. The simulated EM images are compared with the gold standard atom-based ice model approach and shown to be directly comparable. Comparison with experimentally acquired data shows the Gaussian random field ice model produces realistic simulations. The software required has been implemented in the Parakeet software package and the underlying atomic models are available online for use by the wider community.
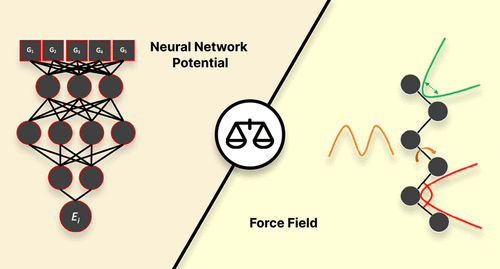
We present a comparative study that evaluates the performance of a machine learning potential (ANI-2x), a conventional force field (GAFF), and an optimally tuned GAFF-like force field in the modeling of a set of 10 γ-fluorohydrins that exhibit a complex interplay between intra- and intermolecular interactions in determining conformer stability. To benchmark the performance of each molecular model, we evaluated their energetic, geometric, and sampling accuracies relative to quantum-mechanical data. This benchmark involved conformational analysis both in the gas phase and chloroform solution. We also assessed the performance of the aforementioned molecular models in estimating nuclear spin–spin coupling constants by comparing their predictions to experimental data available in chloroform. The results and discussion presented in this study demonstrate that ANI-2x tends to predict stronger-than-expected hydrogen bonding and overstabilize global minima and shows problems related to inadequate description of dispersion interactions. Furthermore, while ANI-2x is a viable model for modeling in the gas phase, conventional force fields still play an important role, especially for condensed-phase simulations. Overall, this study highlights the strengths and weaknesses of each model, providing guidelines for the use and future development of force fields and machine learning potentials.
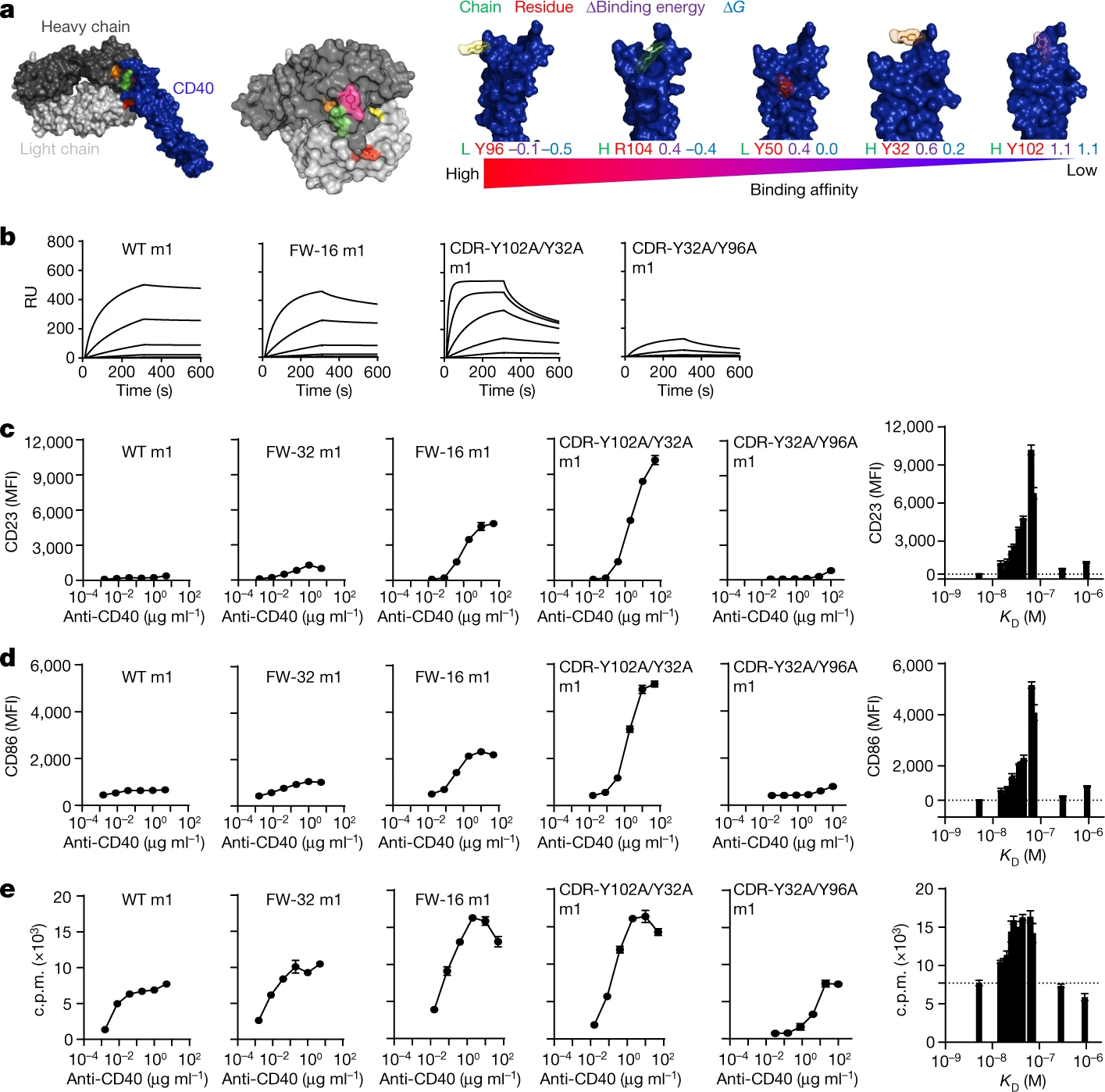
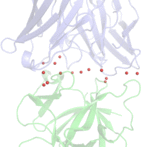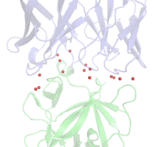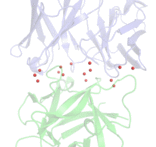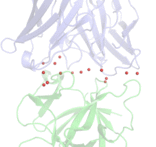
Antibody responses during infection and vaccination typically undergo affinity maturation to achieve high-affinity binding for efficient neutralization of pathogens. Similarly, high affinity is routinely the goal for therapeutic antibody generation. However, in contrast to naturally occurring or direct-targeting therapeutic antibodies, immunomodulatory antibodies, which are designed to modulate receptor signalling, have not been widely examined for their affinity–function relationship. Here we examine three separate immunologically important receptors spanning two receptor superfamilies: CD40, 4-1BB and PD-1. We show that low rather than high affinity delivers greater activity through increased clustering. This approach delivered higher immune cell activation, in vivo T cell expansion and antitumour activity in the case of CD40. Moreover, an inert anti-4-1BB monoclonal antibody was transformed into an agonist. Low-affinity variants of the clinically important antagonistic anti-PD-1 monoclonal antibody nivolumab also mediated more potent signalling and affected T cell activation. These findings reveal a new paradigm for augmenting agonism across diverse receptor families and shed light on the mechanism of antibody-mediated receptor signalling. Such affinity engineering offers a rational, efficient and highly tuneable solution to deliver antibody-mediated receptor activity across a range of potencies suitable for translation to the treatment of human disease.
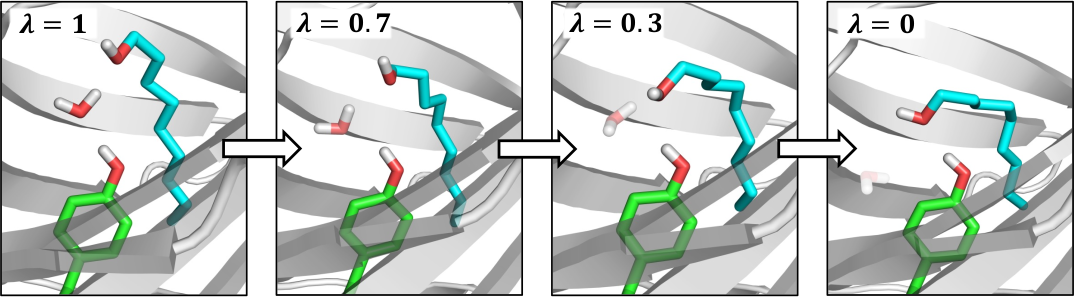
Water molecules play a key role in many biomolecular systems, particularly when bound at protein–ligand interfaces. However, molecular simulation studies on such systems are hampered by the relatively long time scales over which water exchange between a protein and solvent takes place. Grand canonical Monte Carlo (GCMC) is a simulation technique that avoids this issue by attempting the insertion and deletion of water molecules within a given structure. The approach is constrained by low acceptance probabilities for insertions in congested systems, however. To address this issue, here, we combine GCMC with nonequilibium candidate Monte Carlo (NCMC) to yield a method that we refer to as grand canonical nonequilibrium candidate Monte Carlo (GCNCMC), in which the water insertions and deletions are carried out in a gradual, nonequilibrium fashion. We validate this new approach by comparing GCNCMC and GCMC simulations of bulk water and three protein binding sites. We find that not only is the efficiency of the water sampling improved by GCNCMC but that it also results in increased sampling of ligand conformations in a protein binding site, revealing new water-mediated ligand-binding geometries that are not observed using alternative enhanced sampling techniques.