


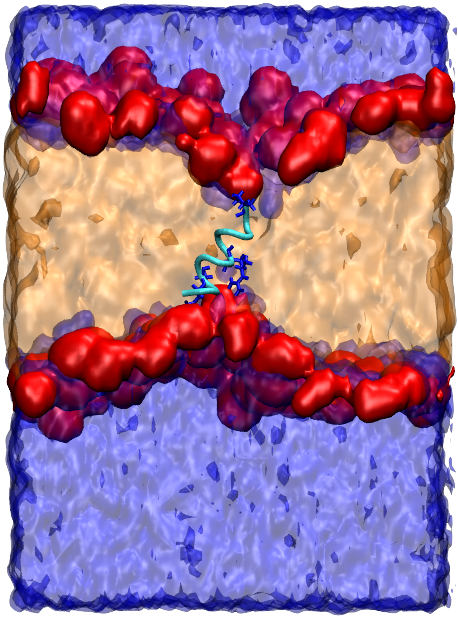
Cell penetrating peptides (CPPs) are short amino acid sequences that are able to translocate into cells without causing significant disruption to the cell membrane. The ability of CPPs to pull large cargo molecules into the cytoplasm makes them attractive as potential intracellular delivery vectors, which are essential for the delivery of biopharmaceutical treatments for intracellular diseases (such as cancer) and cellular imaging agents for diagnostics. Understanding the atomistic interactions and mechanisms of CPPs with cell membranes will lead to the design of more efficient delivery vectors, however applying experimental techniques for this purpose is challenging and the exact mechanisms of many CPPs remain uncharacterised. Molecular simulation offers an approach that can probe the interactions between CPPs and membrane models at the atomistic level, which can provide useful insight that can validate or challenge our experimental understanding of the cell-penetration process.We have benchmarked the use of a number of biomolecular force fields with an enhanced conformational sampling protocol (REST2) in aqueous and membrane-mimicking solvent models for the prediction of environment-specific peptide conformational ensembles. This allowed us to predict the structural transitions that occur in TP2, a known CPP, as it passes through the membrane and enters the cytoplasm. Using the calculated structures of TP2, we also investigated the interactions and free energy profile associated with the translocation process using phospholipid bilayer self assembly and umbrella sampling simulations. This led to the identification of a translocation pathway that involves the formation of an amphipathic TP2 helix upon penetration into the membrane, followed by the formation of a stable transmembrane helix where the cationic peptide residues interact with the polar phosphate headgroups from both sides of the bilayer, and the hydrophobic peptide residues interact with the hydrophobic lipid chains.